Nat Commun:图谱级单细胞数据高效整合算法“scMerge2”,实现跨样本、跨条件分析
时间:2023-08-15 23:35:06 热度:37.1℃ 作者:网络
近年来,大规模单细胞基因和蛋白质图谱分析技术飞速发展,包括单细胞转录组测序(scRNA-seq)、飞行时间质谱流式(CyTOF)和成像质谱流式等。这些技术使科研人员对单个细胞的特性和功能有了深入了解,改变对细胞类型特异性以及生物系统的理解。同时,海量的单细胞数据也推动着不同研究数据图谱的整合。多个大型队列研究的有效整合,有望提供单个研究无法提供的不同条件下细胞的生物学见解,为探索健康和患病细胞提供了有价值的参考。但这类数据和研究需要相应的算法,可对多个数据集进行有效访问和联合解读,以释放单细胞分辨率荟萃分析的力量。
近日,澳大利亚悉尼大学的研究人员在Nature Communications发表了题为“Atlas-scale single-cell multi-sample multi-condition data integration using scMerge2”的文章。研究团队推出了一种名为scMerge2的可扩展算法,能够对图谱规模多样本、多条件单细胞研究进行数据整合,以帮助人们了解不同条件下细胞的生物学特性。经验证,scMerge2能够实现来自多个队列的scRNA-seq数据整合,并揭示来自细胞类型表达的特征。此外,scMerge2还可以消除CyTOF、CITE-seq等试验数据集的可变性,适用于广泛的单细胞分析技术。

文章发表在Nature Communications
scMerge2方法原理如图1所示,其通过对稳定表达的基因进行因子分析和跨数据集的伪复制来整合多个单细胞转录组数据,并增强生物学发现,比如推断细胞发育轨迹。另外,该方法还支持多种整合设置,可实现跨批次、跨数据集和跨物种的发现。
随着多样本、多条件单细胞研究的快速出现和用于集成数据集数量的增加,scMerge2能够解决细胞和研究的可扩展性以及产生分析准备数据(即调整表达矩阵)相关的挑战。scMerge2存在三个关键的创新点:(1)通过分层整合捕获不同研究之间的局部和整体差异;(2)通过伪bulk构造确保计算可扩展性;(3)在每个条件内进行伪复制来捕获来自多个条件的信号。
研究团队将scMerge2与另外两种数据集成方法Seurat、fastMNN做了比较。这两种方法需要严格的合并策略且只允许在每个级别进行成对合并,并以渐进的方式执行批量合并。相比之下,scMerge2提供了更灵活、适应性更强的多级合并结构,其中每个级别可以包含多个批次的多个集合,并且可以使用用户定义的批次标签在每个集合中单独进行批次校正。
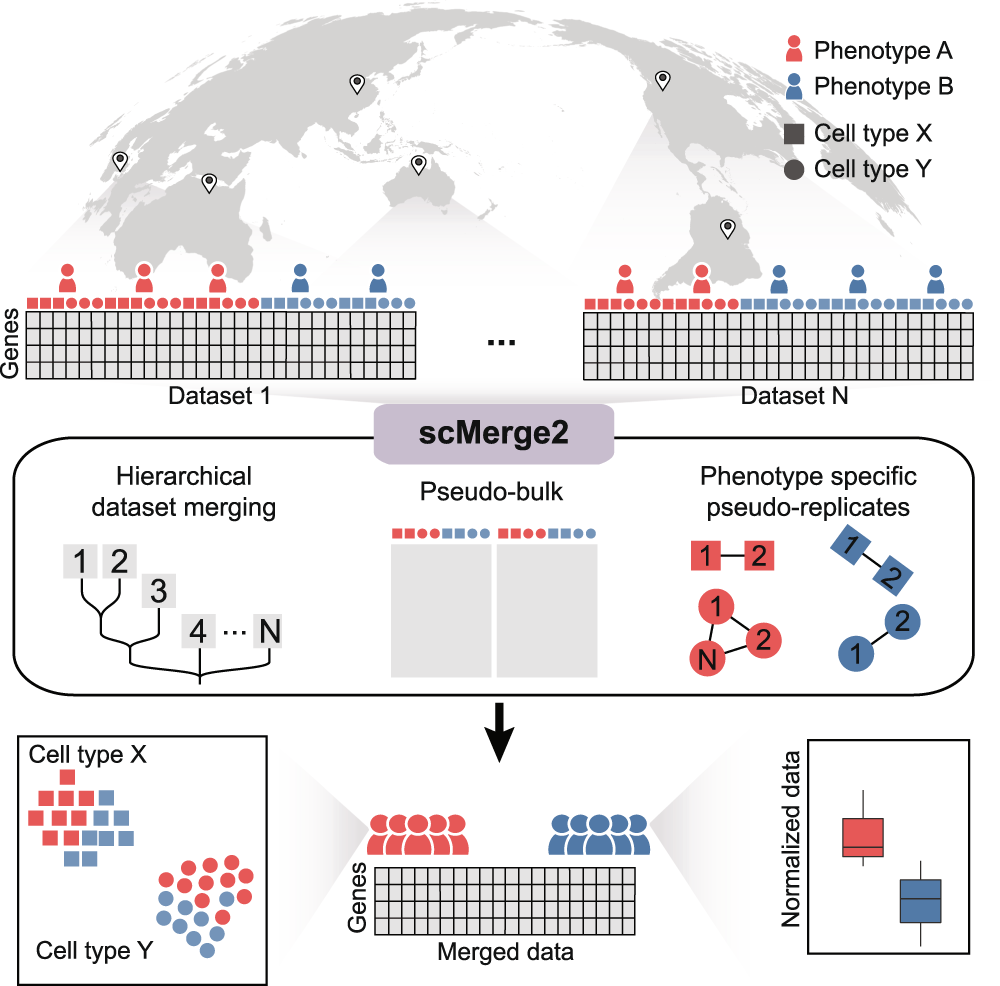
图1. scMerge2概述。
为证明scMerge2分层整合策略的有效性,研究团队将其应用于来自两项COVID-19研究的20万个细胞子集中,比较了两种不同scMerge2设置的性能:scMerge2-h,先进行研究内校正,再进行研究间校正;scMerge2,一次性整合两个数据集(6个批次)(图2)。结果显示,以分层方式整合两项研究可以提高数据整合的性能,特别是在揭示细胞类型信号方面。与其他数据集成方法相比,scMerge2的两种设置在平衡批次效应去除和生物信号保存的方面性能更佳。
研究团队通过改变细胞数量和特征数量衡量了集成方法的可扩展性。结果显示,在所有能够返回调整基因表达矩阵的方法中,scMerge2的计算时间最高效;与fastMNN和scVI相比,scMerge2需要更多的运行内存,但明显少于Seurat。
研究团队通过改变算法的关键调整参数评估了scMerge2的鲁棒性,包括非必要变化因子的数量、伪bulk的数量等。结果显示,虽然算法中的设置不同,但scMerge2始终比其他方法具有更好的性能。上述结果证明了scMerge2在scRNA-seq数据集成中的有效性、实用性和计算效率。
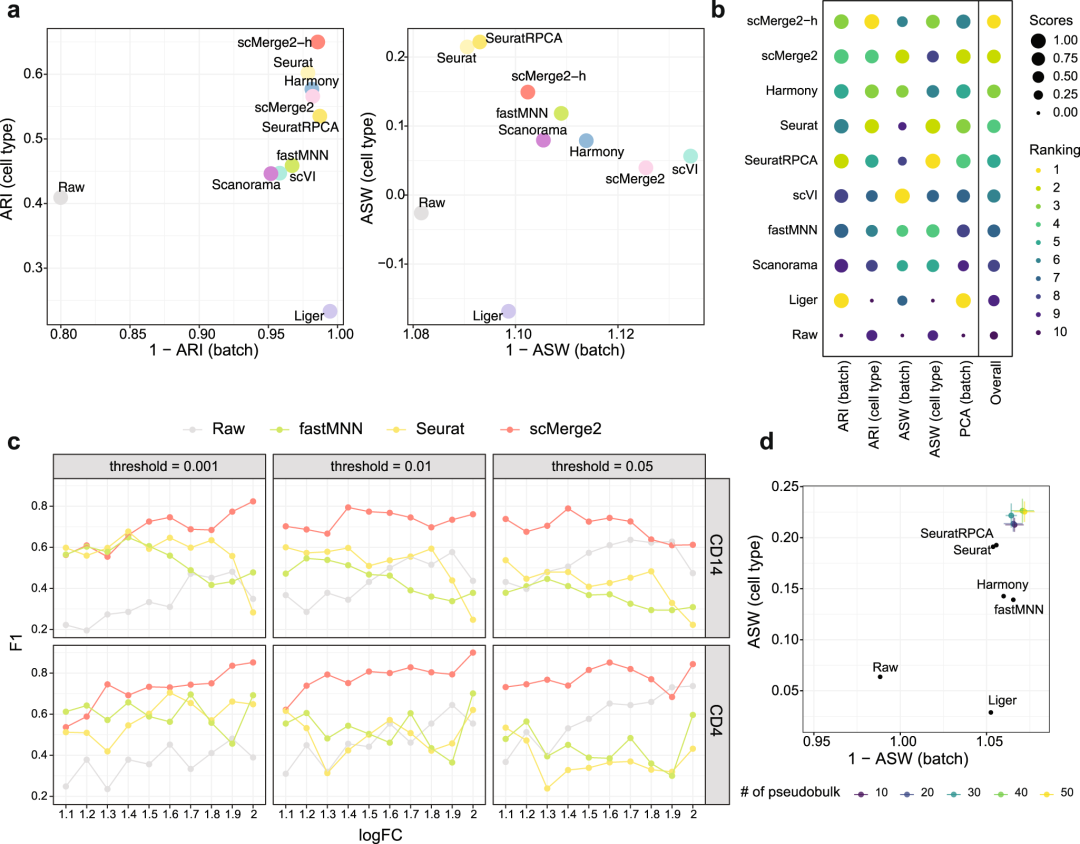
图2. scMerge2的性能优于现有的整合方法。
为证明scMerge2在整合多样本多条件单细胞数据方面的可扩展性,研究团队对COVID-19数据集进行了scMerge-2整合,该数据集包括全球20项研究的1298个PBMC样本(963个个体)中约500万个细胞(图3)。UMAP可视化图显示,scMerge2有效地整合了20项研究,同时保留了多层次的细胞类型信息。按数据集划分的UMAP图进一步表明,scMerge2成功地消除了由数据集引起的非必要变化。
定量评估指标证实了上述观察结果,scMerge2减少了由数据集、操作流程和技术引起的技术层面差异,从而提升了细胞类型识别性能。此外,基于细胞类型特异性表达,scMerge2改善了对COVID-19数据集中疾病严重程度的预测。与原始对数标准化数据相比,使用scMerge2识别细胞类型大大提高了数据中丰度大于1%的所有细胞类型的疾病严重程度预测准确率,平均准确率提高3.2%。
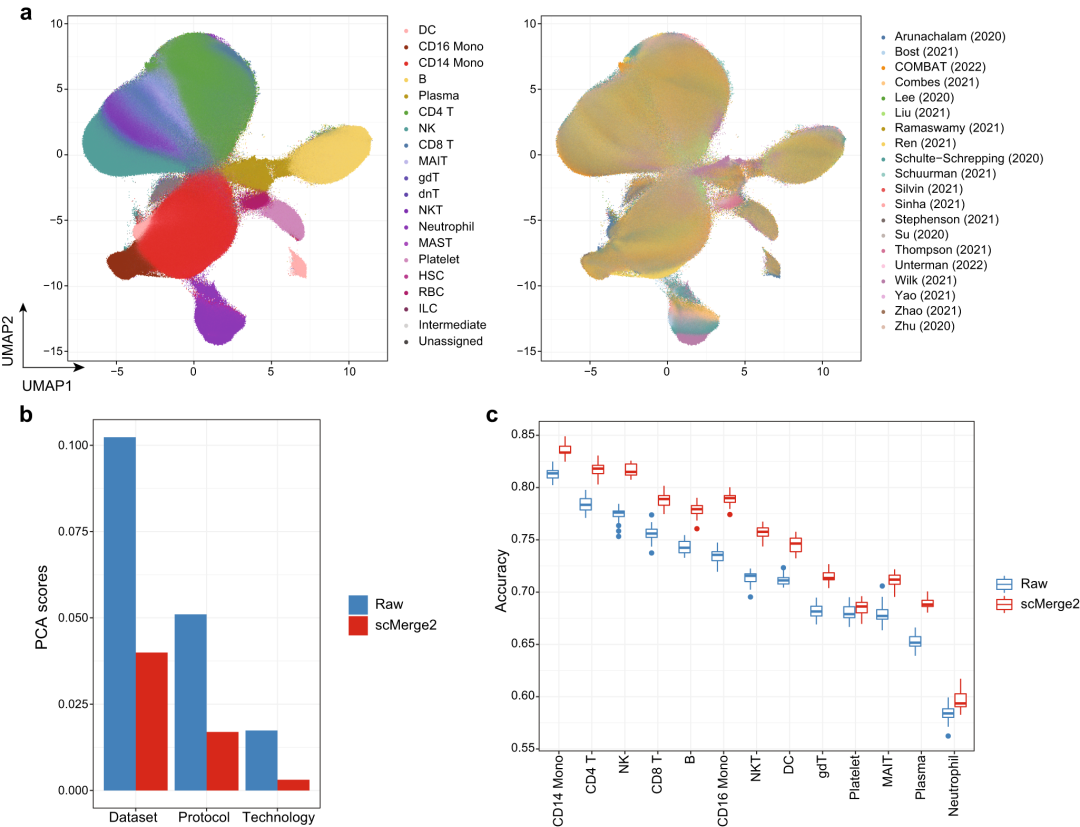
图3. scMerge2可扩展整合500万新冠肺炎PBMC细胞。
研究团队分析了与每种细胞类型的疾病严重程度和时间相关的细胞类型特异性潜在生物过程途径。将疾病严重程度和发病天数作为协变量,对细胞类型特异性pseudo-bulk进行差异表达分析,然后进行基因集富集分析。
结果显示,与疾病严重程度相关的信号通路(包括标志性TNFα信号传导和标志性炎症反应)在大多数细胞类型的重症患者中上调,而GO IL6阳性表达和标志性MTORC1信号传导在中度患者中上调。上述结果表明,利用scMerge2整合多项研究,可采用多种数据分析方法来解决广泛的生物学问题。
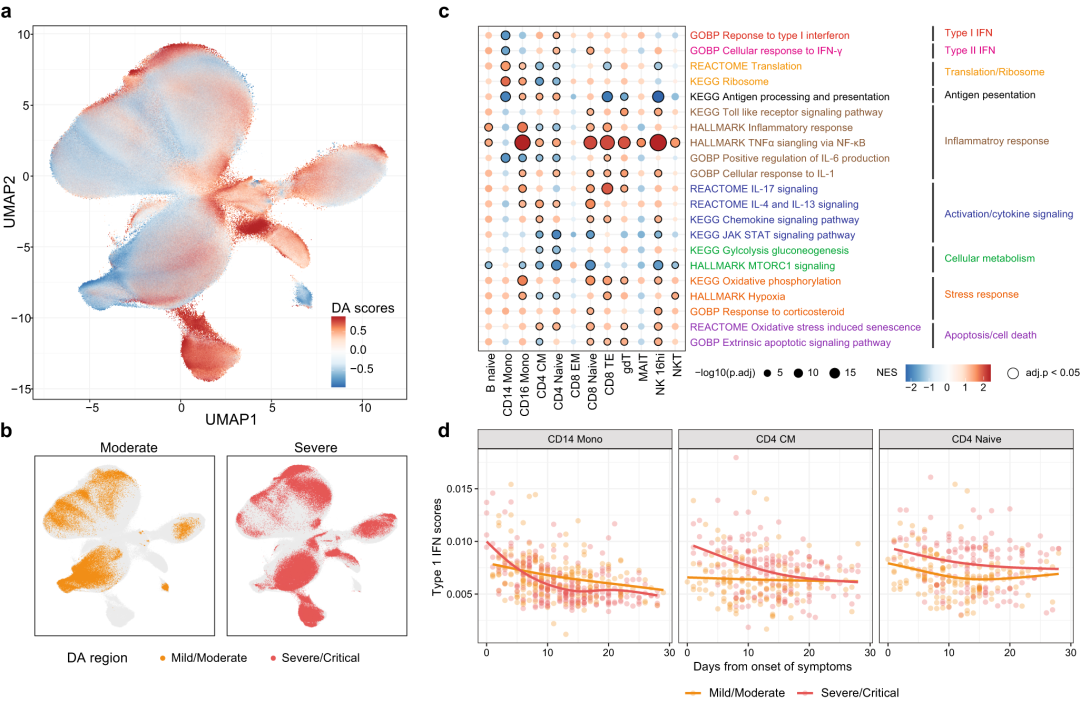
图4. scMerge2支持对多条件数据进行差异细胞状态检测。
scMerge2的主要优势之一是对来自多个生物技术平台数据的通用性,其适用于各种单细胞模态,包括空间分辨模态和多模态。研究团队以两个CyTOF数据集为例,证明了scMerge2可直接应用于其他单细胞单模态数据(图5)。UMAP图显示,与原始数据相比,两个数据集成功整合。值得注意的是,scMerge2还能够揭示仅存在于特定批次的独特细胞类型。
接下来,研究团队证明了scMerge2能够对空间分辨的单细胞数据进行规范化处理,从而更好地识别特定聚类标记的细胞类型。以COVID-19成像质谱(IMC)数据集为例,研究团队发现与原始数据相比,scMerge2调整矩阵提供了更好的聚类结果,与手动细胞类型注释更一致,ARI从0.13增加到0.5。
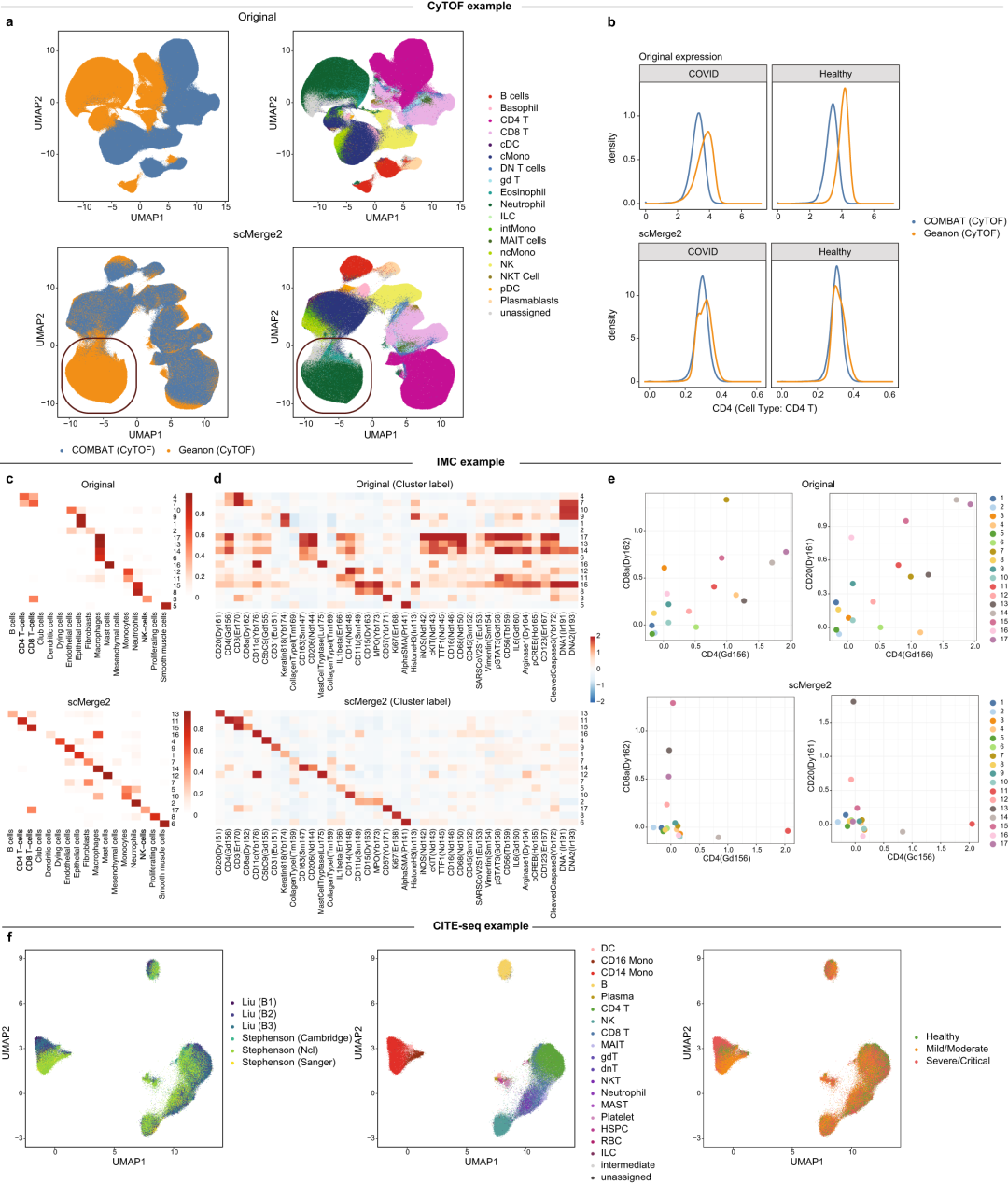
图5. scMerge2适用于其他单细胞平台。
综上所述,研究团队提出了scMerge2方法,能够对大量单细胞数据进行图谱级整合分析,实现有效的下游荟萃分析。该研究揭示,scMerge2可整合来自多种单细胞技术的数百万个细胞,其在所有主要细胞类型的预测准确率方面都有显著提高,并能够检测不同严重程度COVID-19患者的不同细胞状态。随着公共多样本、多条件单细胞研究数量的持续激增,scMerge2有望得到越来越广泛的应用。
参考资料:
Lin, Y., Cao, Y., Willie, E. et al. Atlas-scale single-cell multi-sample multi-condition data integration using scMerge2. Nat Commun 14, 4272 (2023). https://doi.org/10.1038/s41467-023-39923-2

